Docs
Background#
Making libraries for single-cell sequencing can be complex and expensive. With ResolveDNA Whole Genome Amplification Kit we have simplified and provided a robust way to generate single-cell libraries. One way users can ensure that the single-cell library is uniformly amplified with low allelic dropouts, is by first sequencing using “low-pass” or low throughput sequencing of around 2M reads per sample. Data from the “low-pass” is used to estimate the genome coverage if the single-cell libraries were used for high-depth sequencing. Users can then only use the passing libraries for high-depth sequencing.
We provide all ResolveDNA Whole Genome Amplification Kit users ability to assess their single-cell libraries by running BioSkryb’s BJ-DNA-QC pipeline through our analytics platform called BaseJumper. The BJ-DNA-QC pipeline uses low-pass sequencing data and generates several QC metrics that help assess whether the single-cell libraries are ready for high-depth sequencing.
Pipeline Overview#
flowchart LR
%% Colors %%
classDef panel fill:transparent,stroke:#323232,stroke-dasharray:8
classDef black fill:#12294C,stroke:#12294C,stroke-width:2px,color:#fff
classDef blue fill:#20A4F3,stroke:#20A4F3,stroke-width:2px,color:#fff
classDef green fill:#3BCEAC,stroke:#3BCEAC,stroke-width:2px,color:#fff
classDef yellow fill:#ffd166,stroke:#ffd166,stroke-width:2px,color:#fff
classDef pink fill:#ef476f,stroke:#ef476f,stroke-width:2px,color:#fff
classDef orange fill:#f3722c,stroke:#f3722c,stroke-width:2px,color:#fff
classDef red fill:#BB4430,stroke:#BB4430,stroke-width:2px,color:#fff
classDef ming fill:#387780,stroke:#387780,stroke-width:2px,color:#fff
Start((Start)):::black --fastq--> Trimming[Subsample Reads <br/> <br/> Trim Reads]:::blue
subgraph Preprocess
Trimming
end
subgraph Map
direction TB
Trimming --> Alignment[Align Reads <br/> <br/> Remove Duplicates]:::green
end
subgraph Evaluate
Trimming --> M_FastQC[Read Metrics]:::pink
Alignment --> M_Sentieon[Alignment Metrics <br/><br/> GC Metrics <br/><br/> Insert Size Metrics <br/><br/> Coverage Metrics]:::pink
Alignment --> M_Preseq[Library Complexity Metrics]:::pink
Alignment --> M_CNV[CNV]:::pink
end
subgraph Report
M_FastQC --> Mqc[MultiQC Report]:::orange
M_Sentieon --> Mqc
M_Preseq --> Mqc
end
Mqc --> End((End)):::black
Preprocess:::panel
Map:::panel
Evaluate:::panel
Report:::panelFollowing are the steps and tools that pipeline uses to perform the analyses:
-
Subsample the reads to 2 million using
SEQTK SAMPLEto compare metrics across samples -
Evaluate sequencing quality control using
FASTPand trim/clip reads -
Map reads to reference genome using
SENTIEON BWA MEM -
Remove duplicate reads using
SENTIEON DRIVER LOCUSCOLLECTORandSENTIEON DRIVER DEDUP -
Evaluate metrics using
SENTIEON DRIVER METRICSwhich includes Alignment, GC Bias, Insert Size, and Coverage metrics -
Evaluate the BAM quality control using
QUALIMAP BAMQC -
Evaluate the library complexity using
PRESEQ BAM2MRandPRESEQ GC EXTRAP -
Evaluate the CNV using a custom
Ginkgoimpelmentation -
Aggregate the metrics across biosamples and tools to create overall pipeline statistics summary using
MULTIQC
Pipeline Parameters#
| Parameter Name | Options |
Description |
|---|---|---|
| Read Length | 50 75 (default) 100 150 |
Read length preference for each sample can be made here and used for sequencing |
| Read Sampling | 1000 1000000 2000000 (default) |
Number of reads to sample. 1M paired reads is equivalent to 2M individual reads |
| Genome | GRCh38 (default) GRCm38 GRCm39 |
Reference genome to use for alignment |
Module Parameters#
| Module | Parameter Name | Options | Description |
|---|---|---|---|
| FastQC | (default) | FastQC performs qc checks on your raw sequence data. | |
| Qualimap | (default) | Qualimap module evaluates the quality of the alignment data. | |
| CNV | (default) | CNV module evaluates the Copy Number Variation. |
Output Files#
Output Directory/File |
Notes |
|---|---|
multiqc/ |
This section includes output files containing metrics from various tools to create a MultiQC report. MultiQC Report Example |
primary_analyses/ metrics/ |
Metrics output from Fastp, Kraken2, and/or FASTQC if those modules were selected to run the analyses for each biosample. |
secondary_analyses/ alignment/ metrics/ |
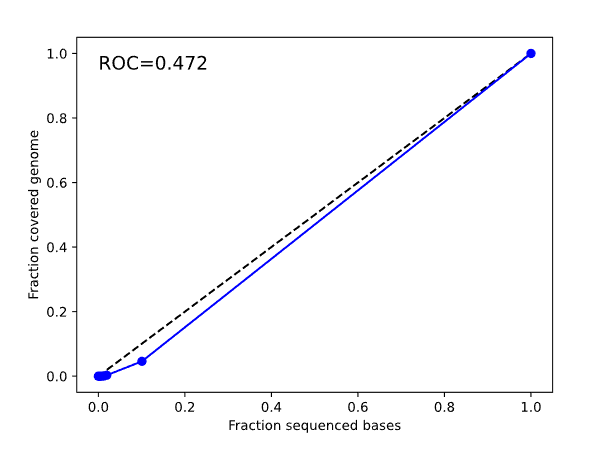
alignment/Biosample level output containing aligned reads and index file on subsample reads. metrics/ Metrics output from secondary analyses - Alignment, GC bias, Insert Size, Coverage, and library complexity metrics. The section includes outputs from the Bam Lorenz coverage tool containing information about coverage using lorenz curve estimation in order to look at uniformity across the genome. The *-pipeline_all_metrics_mqc.txt contains metrics from the All Metrics section of the MultiQC report found in BaseJumper. Bam lorenz curve Example |
tertiary_analyses/ cnv_ginkgo/ |
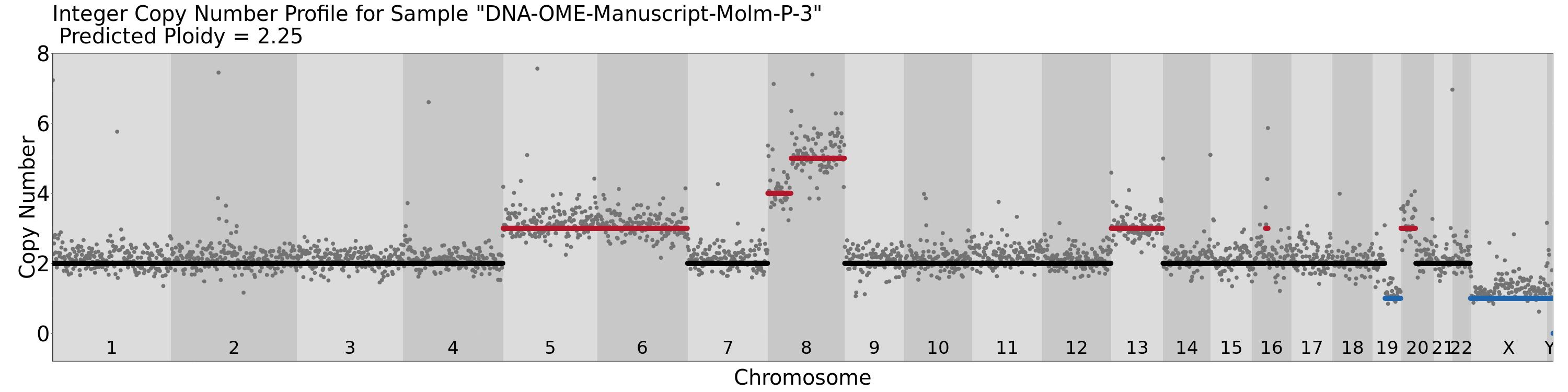
Biosample level output from Ginkgo. CNV profile Example |
execution_info/ |
This section includes execution information regarding the pipeline run. |
{kind=link}
{kind=link}