BJ-SV
Background#
BJ-SV is a pipeline that detects structural variants in BaseJumper using Manta and Expansion Hunter tools. The Manta tool is used to detect structural variants and indels. The Expansion Hunter tool is used to detect repetition regions in the genome. Both of these tools combined together are able to produce better structural variants performance. Structural Variants performance is essential for understanding in order to improve and understand aberrations in your data. BJ-SV is a tertiary pipeline in BaseJumper and is dependent on the secondary BJ-WGS pipeline. Once the BJ-WGS pipeline completes successfully, the prerequisites are met to launch the BJ-SV pipeline.
Pipeline Overview#
Variant Calling - This step performs calling of germline, joint and copy number variants. The inputs are deduplicated (or deduplicated and compressed) .bam files, various variant databases, machine learning model and targeted .bed file. Outputs are variants described in .vcf files and copy number information in .tsv file and various plots.
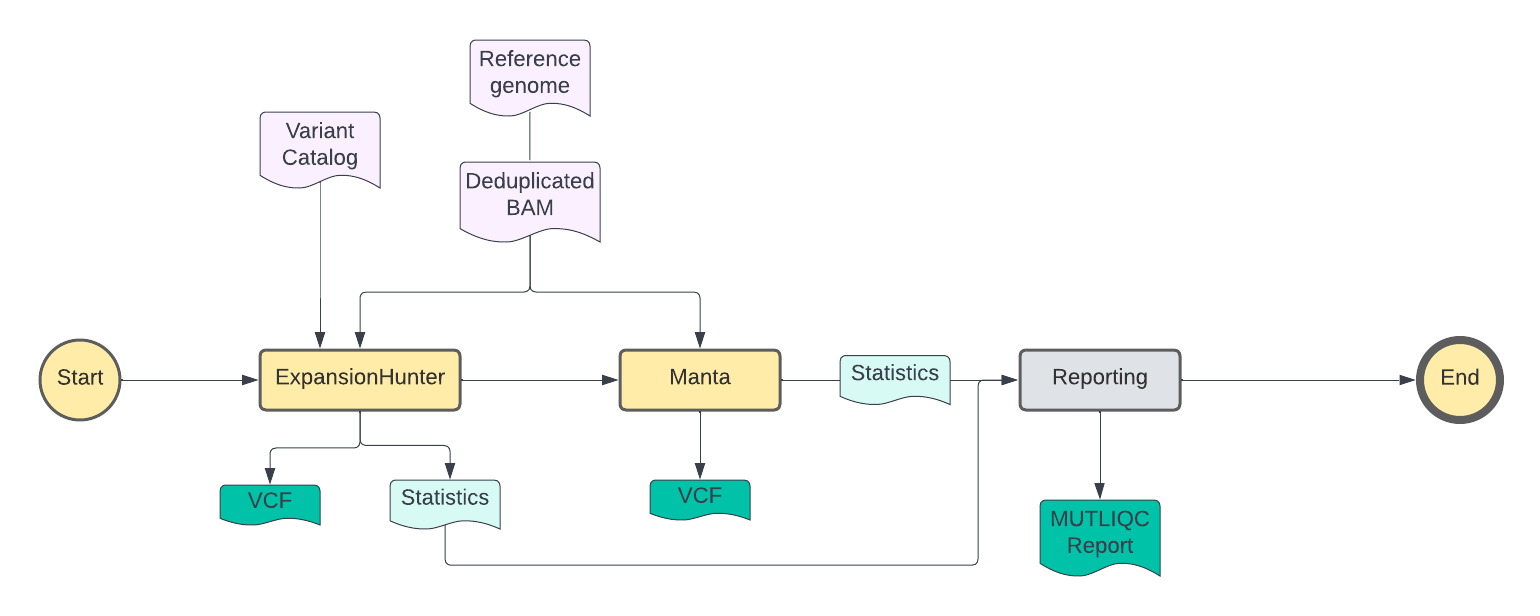
Structural Variants - Manta and Expansion Hunter:
Exapnsion Hunter
This tool is used to detect repetition regions in the genome. This takes the WGS .bam files from DEDUP and uses them to find the size of the Short Tadem Repeats(STR) in reference to the size of the read length outputs a .vcf file. The expasion of these read lengths based on STR means that there is a biological disorder and can causes diseases such as Huntington and Fredreich ataxia (FRDA). More information on Expansion Hunter can be found here
Manta
This tool is used to detect structural variants and indels. This takes the WGS .bam files from the DEDUP and uses them to score variants in order to perform germline analyses creating an output file of a .vcf. These variant scores are produced based on the identification of the pair or split read regions in the genome of a single cell. 2 Manta has known limitations and this is provided in the next section below. More information on Manta can be found here
Parameters/Modules#
| Module | Parameter Name | Options | Description |
|---|---|---|---|
| Genome | GRCh38(default) | Options for choosing reference genome being used for analyses. | |
| SVs | (default) | Calls structural variants with Manta tool. | |
| STRs | (default) | Calls short tandem repeats with Expansion Hunter tool. |
Key Components#
Variant Calling - This step performs calling of germline, joint and copy number variants. The inputs are deduplicated (or deduplicated and compressed) .bam files, various variant databases, machine learning model and targeted .bed file. Outputs are variants described in .vcf files and copy number information in .tsv file and various plots.
Structural Variants - Manta and Expansion Hunter:
Exapnsion Hunter
This tool is used to detect repetition regions in the genome. This takes the WGS .bam files from DEDUP and uses them to find the size of the Short Tadem Repeats(STR) in reference to the size of the read length outputs a .vcf file. The expasion of these read lengths based on STR means that there is a biological disorder and can causes diseases such as Huntington and Fredreich ataxia (FRDA). 1 More information on Expansion Hunter can be found here
Manta
This tool is used to detect structural variants and indels. This takes the WGS .bam files from the DEDUP and uses them to score variants in order to perform germline analyses creating an output file of a .vcf. These variant scores are produced based on the identification of the pair or split read regions in the genome of a single cell. 2 Manta has known limitations and this is provided in the next section below. More information on Manta can be found here
Known Limitations#
The section talks about all of the known limitations found with the Manta tool. 2
Manta should not be able to detect the following variant types:
- The limiting size is not tested, but in theory detection falls off below ~200 bases. Micro-inversions might be detected indirectly when insertion and deletion variants are combined.
- The maximum fully-assembled insertion size should be similar to approximately double the read-pair fragment size, however, the power to fully assemble the insertion should fall off to impractical levels before this size.
More general repeat-based limitations exist for all variant types:
- Power to assemble variants to breakend resolution falls to zero as breakend repeat length approaches the read size.
- Power to detect any breakend falls to (nearly) zero as the breakend repeat length approaches the fragment size.
The following limitations exist on the input BAM or CRAM files provided to Manta:
- Alignments must not have an unknown read sequence (SEQ="*")
- Alignments must not contain the "=" character in the SEQ field.
- Alignments must not use the sequence match/mismatch ("="/"X") CIGAR notation RG (read group) tags in the alignment records are ignored -- each file will be treated as representing one sample.
- Alignments that have basecall quality values larger than 70, will be rejected (these are not supported on the assumption that this indicates an offset error)
Note
Manta can be unstable due to input of .bam files. The fraction of 'Rp' among total high-confidence read pairs needs to be more than 0.9 to determine consensus pair orientation.
Manta classifies novel DNA-adjacencies, it does not infer the higher level constructs implied by the classification. For instance, a variant marked as a deletion by manta indicates an intrachromosomal translocation with a deletion-like breakend pattern, however there is no test of depth, b-allele frequency or intersecting adjacencies to directly infer the SV type.
Manta does detect and report very large insertions when the breakend signature of such an event is found, even though the inserted sequence cannot be fully assembled.
Manta also requires a reference sequence in fasta format. This must the reference used for mapping the input alignment files. The reference has to include a samtools/htslib-style index in a file named to match the input fasta with an additional '.fai' file extension.
Output files#
Output Directory/File |
Notes |
|---|---|
tertiary_analyses/ manta/ <BIOSAMPLE_NAME>/ <BIOSAMPLE_NAME>.manta.candidateSV.vcf.gz <BIOSAMPLE_NAME>.manta.candidateSmallIndels.vcf.gz <BIOSAMPLE_NAME>.manta.diploidSV.vcf.gz tertiary_analyses/ expansion_hunter/ <BIOSAMPLE_NAME>/<BIOSAMPLE_NAME>.vcf.gz<BIOSAMPLE_NAME>.json |
This section includes the output files for the Variant calling. The Manta output file is to show location and position of indels like insertions and with or not that insertion was fully assembled or not. INS represents insertion and the LEFT_SVINSSEQ and RIGHT_SVINSSEQ is used to show on both ends of the sequence. Another type of output file that you may see is a reciprocal inversion which shows breakends in the inversion. Anxample of these two output files can be found in the Outfile file example section and more descritpion of differnt symbols from the Manta tool can be found in the Output file key for Manta section. The output file from the Expansion Hunter tool and shows the position and location of a chromsome where the number of Tadem Repeats occur. For example. In this file, it shows that on chrosome 9 each allele has a different set of repeat units. <STR> stands for Short Tadem Repeats and the number that comes after it is the # of repeats. so <STR2> means 2 repeats on allele 1 and <STR349> means 349 repeats on allele 2 with the sequence RU: GGCCCC. The REF = 3 means that the repeat extends 3 repeat units. SPANNING is used to decided if the legnth of an allel determine based on the spanning reads or INREPEAT which is used for deciding if the lenght of an allele size is determined based on the in-repeat reads. An Example of this file output is shown in the Output file example section |