BJ-TMB
Background#
BJ-TMB is a Tumor Mutational Burden (TMB) pipeline in BaseJumper. TMB is used by identifying all of the somatic exonic mutations in a tumor to create Biomarkers that will be used as predictions for therapeutics targets. Some studies have used Whole Exome Sequencing (WES) to look at these results. It was recently shown that TMB can also be used from target sequencing gene panels which seems to be more cost effective. There are different types of mutations used to look at TMB. These can be non-synonymous or synonymous. TMB uses TMBleR to quantify differences in variant filtering (e.g. synonymous, known cancer mutations, by VAF) and to easily plot estimated TMB values and look at the differences between certain methods or gene panels.The figure below shows the graphical representation of how this pipeline works. The graphic and more information on this pipeline can be found here1
Pipeline Overview#
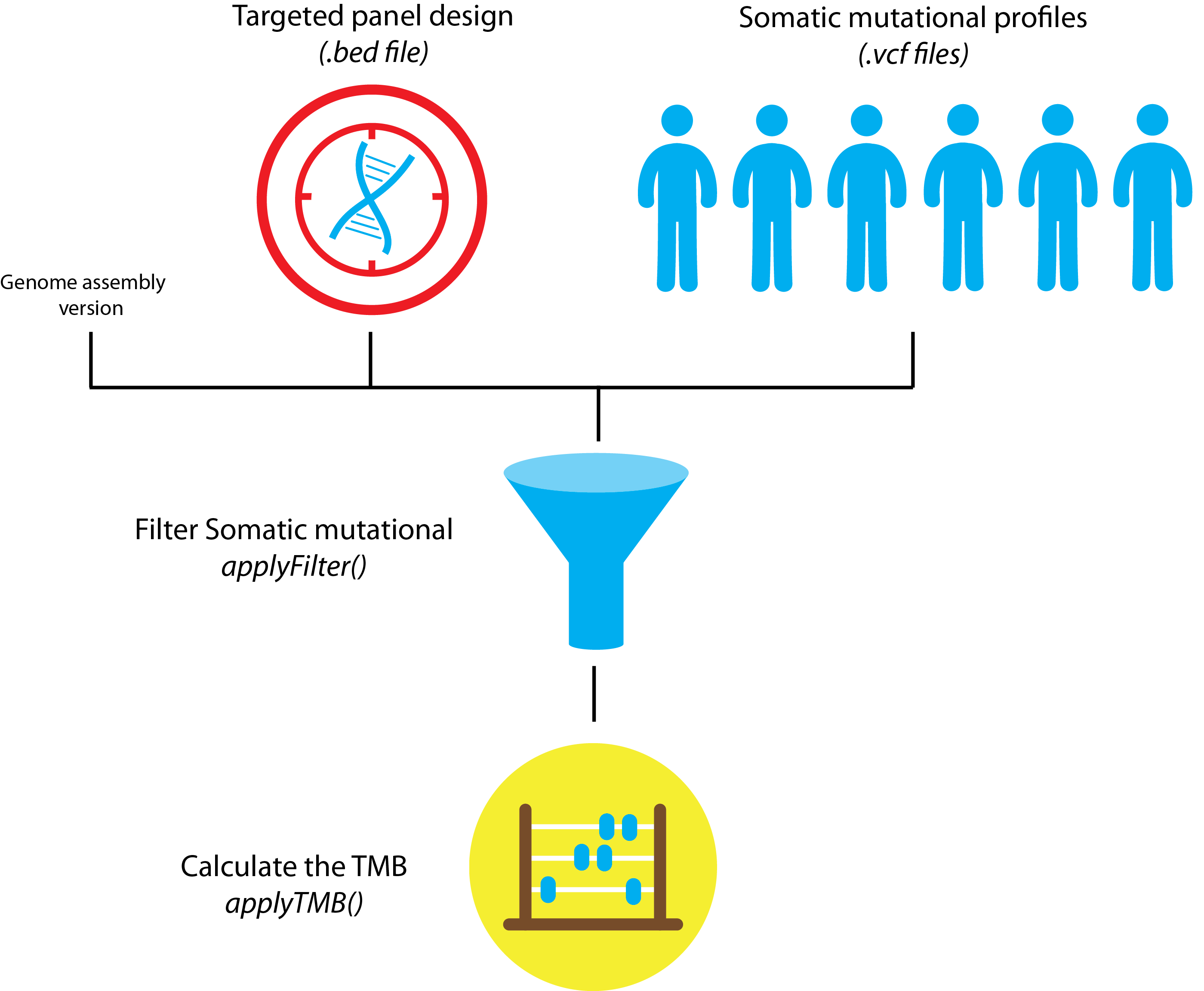
The current pipeline uses only one TMB estimation tool: TMBleR. More about TMBleR tool itself can be found on the following link. Once TMB estimation is completed, MultiQC report will be generated, like shown on the diagram below.
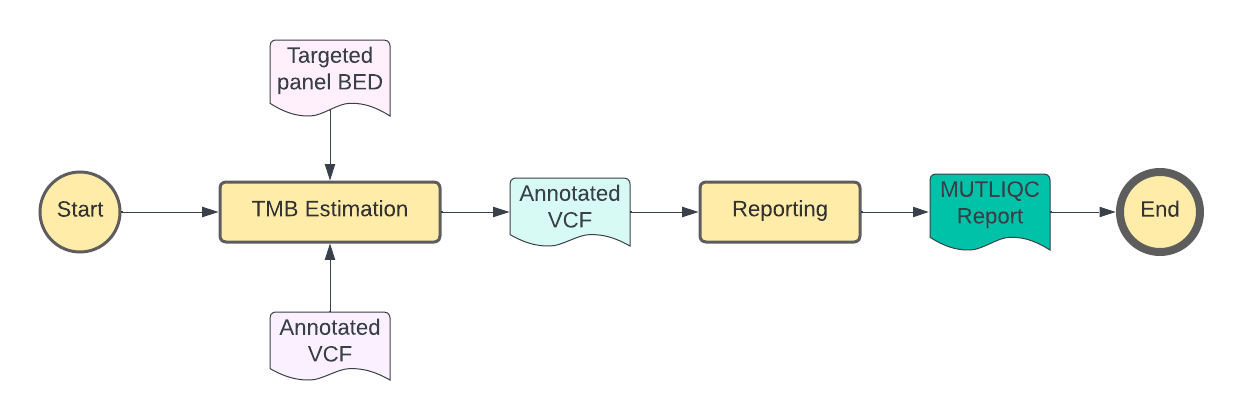
Inputs#
There are 3 inputs required to perform the analysis1
Parameters/Modules#
| Module | Parameter Name | Options | Description |
|---|---|---|---|
| Genome | GRCh38(default) | Options for choosing reference genome being used for analyses. | |
| TMB | (default) | Initiates Tumor Burden Mutational pipeline | |
| TMBleR | (default) | Calls Tumor Burden Estimation tool. |
Output files#
Output Directory/File |
Notes |
|---|---|
tertiary_analyses/ tmb/ <BIOSAMPLE_NAME>/ <BIOSAMPLE_NAME>_TMBquant.tsv |
This section contains TMB Estimation per biosample. TSV outputs are per-sample (they contain information about only one biosample), but contain additional column Design, which indicates which target .bed file was used to calculate TMB. Since this is currently non-modifiable parameter and since it is the same for all runs, this one is not displayed in MULTIQC summary table. 1 |
tertiary_analyses/ multiqc/ multiqc_report.html/ |
This section contains TMB Estimationsummarization for all samples in batch. Results will be diplayed in multiqc_report.html in the tabular form, where one row is dedicated to one biosample1. The table contains following columns: Sample: biosample name Sequencing Size: size of the sequenced genomic space in Mbp Total Number of Mutations: total number of mutations per targeted region per Mb TMB per Mb: estimated TMB per Mb Filters: which filters were used in estimation process. For example, vaf=0NoCancerMutsNosynonymous means:variant allele frequency = 0.0> The options that you will get under the Filter field are: NoCancer: means that variants which can be found in COSMIC database will be ignored. Nosynonymous: means that mutations which change the protein sequences are ignored. |
execution_info/execution_report.htmlexecution_timeline.html |
This section includes execution information from the previous pipeline run. |
Output file Examples#
<BIOSAMPLE_NAME>_TMBquant.tsv
| Sample | Design | Filter | Sequencing_Size | Tot_Number_Mutations | TMB_per_Mb |
|---|---|---|---|---|---|
| Horizon5 | ExamplePanel_GeneIDs | vaf=0NoCancerMuts | 1.861301 | 189 | 101.54 |
| HorizonFFPEmild | ExamplePanel_GeneIDs | vaf=0NoCancerMuts | 1.861301 | 182 | 97.78 |
| Horizon5 | ExamplePanel_GeneIDs | vaf=0NoCancerMutsNosynonymous | 1.861301 | 98 | 52.65 |
| HorizonFFPEmild | ExamplePanel_GeneIDs | vaf=0NoCancerMutsNosynonymous | 1.861301 | 97 | 52.11 |
| Horizon5 | ExamplePanel_GeneIDs | vaf=0Nosynonymous | 1.861301 | 104 | 55.87 |
Reference#
- Fancello, Laura, et al. “TMBleR, a Bioinformatic Tool to Optimize TMB Estimation and Predictive Power.” Bioinformatics (Oxford, England), Dec. 2021, p. btab836. PubMed, https://doi.org/10.1093/bioinformatics/btab836.